
Mukunda Mandal
Friday, December 8, 2023
Mechanistic Insights into Radical Formation and Functionalization in Copper/*N*-Fluorobenzenesulfonimide Radical-Relay Reactions
TOC Graphic
Published In
Authors
Mukunda Mandal,‡* Joshua A. Buss,‡ Si-Jie Chen, Christopher J. Cramer and Shannon S Stahl* (‡Contributed Equally; *Corresponding Author)
Citation
Mandal, M.; Buss, J. A.; Chen, S.-J.; Cramer, C. J.; Stahl, S. S. Mechanistic Insights into Radical Formation and Functionalization in Copper/N-Fluorobenzenesulfonimide Radical-Relay Reactions. Chem. Sci. 2024, 15, 1364.
Express Summary
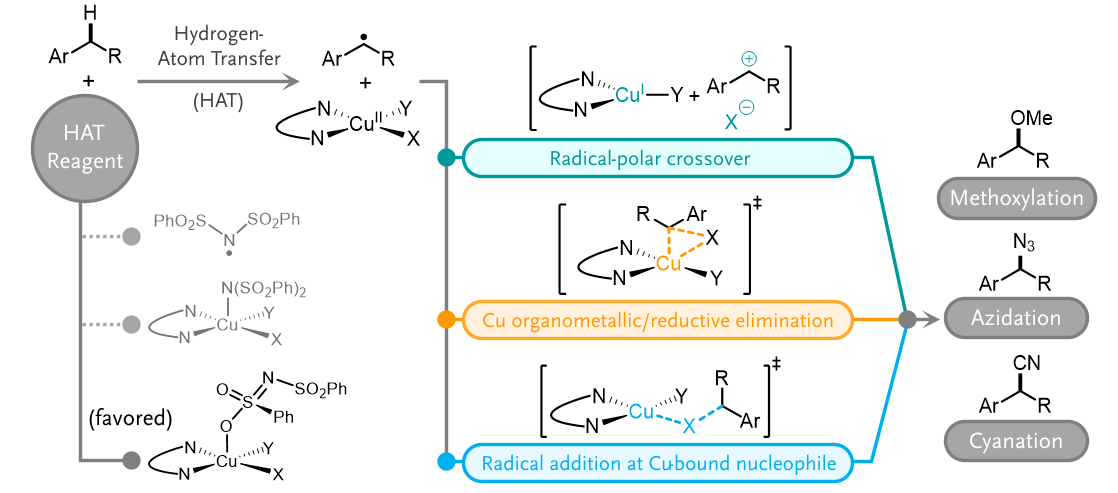
- In Cu-catalyzed C(sp3)–H functionalization with NFSI, the Cu-bound •NSI radical is the active HAT reagent and selectively activates benzylic C–H bonds over 3° C–H bonds (superior vs. other HAT reagents like •OtBu and •Cl).
- The radical functionalization pathway depends on the Cu-bound nucleophile, with options including radical-polar crossover, reductive elimination from a CuIII organometallic complex, and radical addition to a Cu-bound ligand.
- Comparing 3 routes for transforming benzylic C–H bonds into their respective cations revealed that HAT/ET (relevant to Cu/NFSI chemistry) provides a wider substrate scope than hydride transfer (relevant in high-potential quinones) and sequential ET/PT/ET (employed in photoredox catalysis).
Abstract
Copper-catalysed radical-relay reactions that employ N-fluorobenzenesulfonamide (NFSI) as the oxidant have emerged as highly effective methods for C(sp3)–H functionalization. Herein, computational studies are paired with experimental data to investigate a series of mechanistic features of these reactions, with a focus on issues related to site-selectivity, enantioselectivity, and C–H substrate scope. (1) The full reaction energetics of enantioselective benzylic C–H cyanation are probed, and an adduct between Cu and the N-sulfonimidyl radical (•NSI) is implicated as the species that promotes hydrogen-atom transfer (HAT) from the C–H substrate. (2) Benzylic versus 3° C–H site-selectivity is compared with different HAT reagents: Cu/•NSI, •OtBu, and Cl•, and the data provide insights into the high selectivity for benzylic C–H bonds in Cu/NFSI-catalyzed C–H functionalization reactions. (3) The energetics of three radical functionalization pathways are compared, including radical-polar crossover (RPC) to generate a carbocation intermediate, reductive elimination from a formal CuIII organometallic complex, and radical addition to Cu-bound ligand. The preferred mechanism is shown to depend on the ligands bound to copper. (4) Finally, the energetics of three different pathways that convert benzylic C–H bonds into benzylic cations are compared, including HAT/ET (ET = electron transfer), relevant to the RPC mechanism with Cu/NFSI; hydride transfer, involved in reactions with high-potential quinones; and sequential ET/PT/ET (PT = proton transfer), involved in catalytic photoredox reactions. Collectively, the results provide mechanistic insights that establish a foundation for further advances in radical-relay C–H functionalization reactions.
This Article Contains These Key Ideas
- Idea-01.
- Hypothesis-01.
- Proof-01.